
With the collaboration of Dr. Aleks Reinhardt (Department of Chemistry, University of Cambridge), the work of Xiaoliang Tang et al,Numerical calculation of free-energy barriers for entangled polymer nucleation, has been published in The Journal of Chemical Physics.
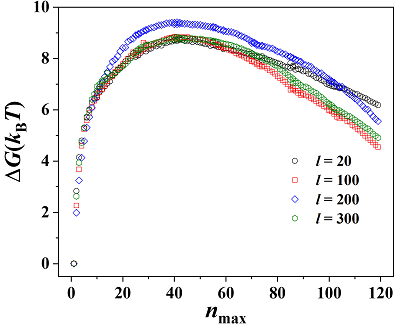
The crystallization of entangled polymers from their melt is investigated using computer simulation with a coarse-grained model. Using hybrid Monte Carlo simulations enables us to probe the behavior of long polymer chains. We identify solid-like beads with a centrosymmetry local order parameter and compute the nucleation free-energy barrier at relatively high supercooling with adaptive-bias windowed umbrella sampling. Our results demonstrate that the critical nucleus sizes and the heights of free-energy barriers do not significantly depend on the molecular weight of the polymer; however, the nucleation rate decreases with increasing molecular weight. Moreover, an analysis of the composition of the critical nucleus suggests that intra-molecular growth of the nucleated cluster does not contribute significantly to crystallization for this system.
We acknowledge financial support from the National Key R&D Program of China (2016YFB0302500), the National Natural Science Foundation of China (51633009), and the Royal Society Newton Mobility Grant MBAG/240 RG82754. We thank Prof. Daan Frenkel and Prof. Chuanfu Luo for fruitful discussions.
Related links: https://aip.scitation.org/doi/full/10.1063/5.0009716, https://arxiv.org/abs/2006.06450